
Research
Our lab is broadly interested in how variation in human genomes leads to the diverse array of phenotypes observed across evolution, diseases, and traits.
We’re a multi-disciplinary team with a variety of backgrounds including genomics, math, biochemistry, machine-learning, and population genetics. We make and apply new technologies to answer a fundamental question remaining in biology: “how do genetic changes lead to functional changes at the molecular, cellular, and phenotypic level?” We’re especially interested in understanding the role of non-coding regulatory elements in the genome, with a special focus on variation within them. We start with genomic signals of positive selection or disease and use a variety of systems to functionally characterize genetic variants underlying those signals. We’re looking for folks to join our team so reach out if you’re interested in human evolution, building new genomic tools, or analyzing complex data. Beyond cool science, we’re a supportive team dedicated to expanding diversity and equity in science.
Current Projects
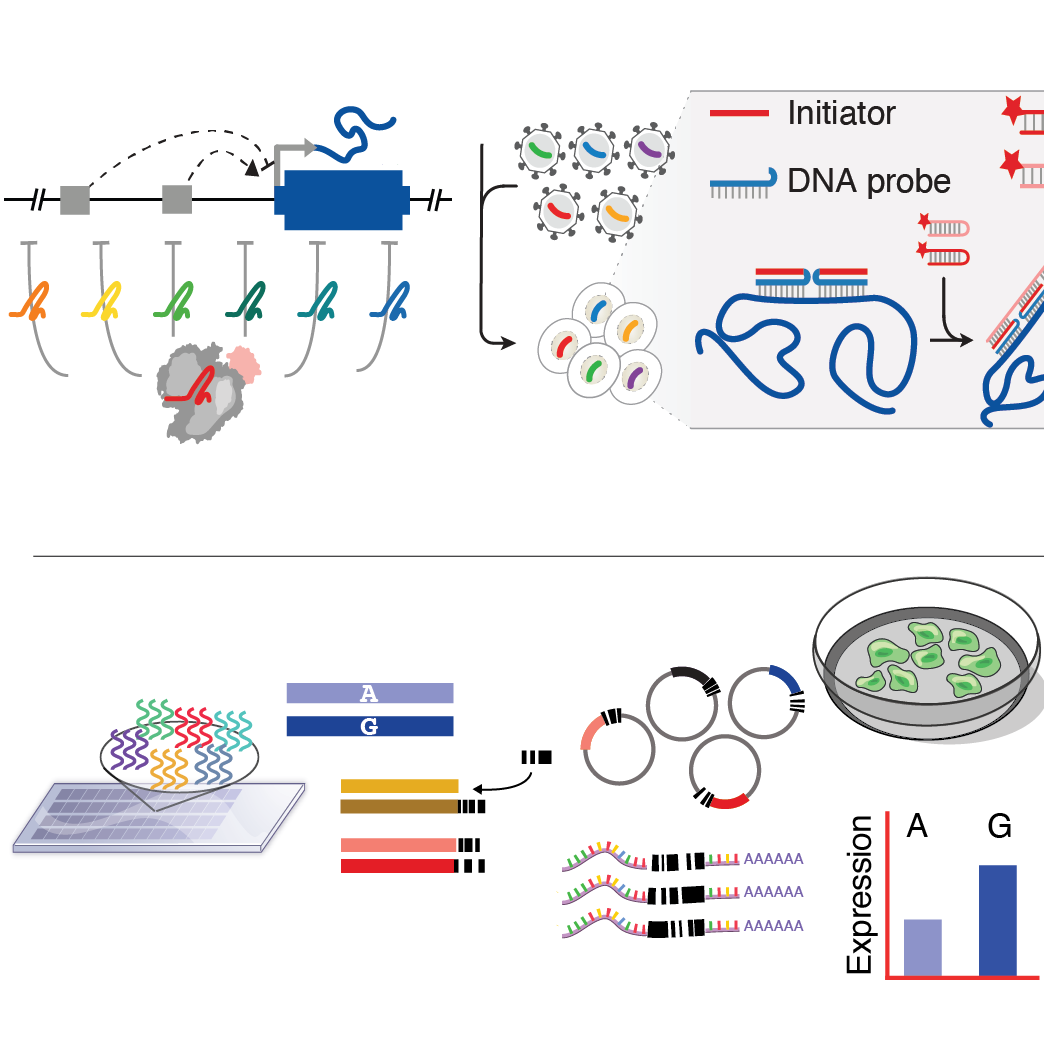
Tools to interpret regulatory variation
We build and apply high-throughput tools to study the functions of cis-regulatory elements (CREs) in the genome. We aim to answer three main questions: 1) which CREs are actually functional in a cell? 2) which gene(s) do they regulate? and 3) how does variation in CREs impact their function? We’ve built tools like HCR-FlowFISH, a CRISPR perturbation strategy to directly characterize CREs, along with CASA, a Bayesian CRISPR analysis strategy. We’ve also applied the Massively Parallel Reporter Assay (MPRA) to understand how variants impact the function of CREs, and worked to expand this approach to other types of non-coding elements, including variation in 3’ UTRs.
We work with the Sabeti and Tewhey labs in their efforts as an ENCODE Functional Characterization Center to directly characterize, rather than map, CREs in the genome. Steve co-leads the CRISPR working group, which is focused on developing best practices and meta-analyses of CRISPR non-coding screens. More broadly, we’ve used our CRE characterization tools to identify functional, causal variants in a wide variety of traits such as lipid metabolism and infectious disease.

ENCODE functional characterization
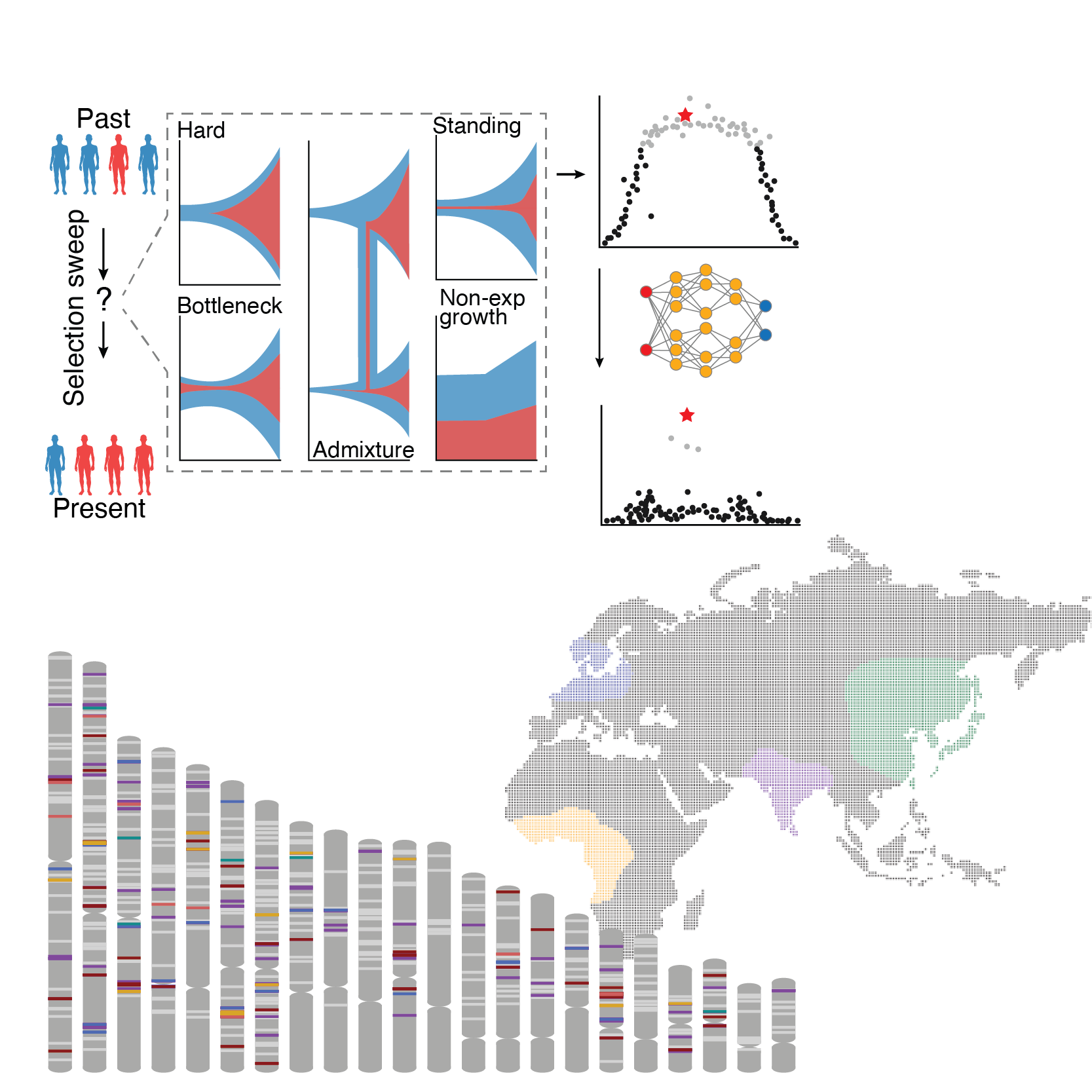
Investigating positive human selection
We’re interested advancing a deep molecular understanding of how positive evolutionary selection has impacted our genomes, specifically how ancient selection impacts health and traits in modern global populations. We’ve built novel computational tools to better model human demographic histories from the 1000 Genomes dataset. We’ve also introduced DeepSweep, a machine learning algorithm to identify and detect positive selection in the human genome with vastly improved accuracy. As most of these selected variants are in non-coding regions of the genome, we applied our CRE characterization tools to link variants to function.
Beyond new functional characterization tools for regulatory variants, more accurate models of regulatory grammar - the code and rulesets underlying the complex logic of cis-regulatory elements - would greatly accelerate our interpretation of the genome. Our lab intends to build new datasets of comprehensively characterized trait-associated regulatory elements, and then use these high-dimensional data to improve our models of CREs.

Learning Regulatory Grammar

Characterizing species-specific variation
Our lab is also interested human-specific sequence changes that may be important for human evolution. As the vast majority of these changes are non-coding, our tools are well positioned to understand the functions of genomic changes between species. We’ve applied the MPRA to characterize all small human Conserved Deletions (hCONDELs) - likely functional sequences conserved in mammals that are deleted only in the human lineage. Understanding the phenotypic consequences of these changes is a major goal of the lab.